")
基本介紹
- 中文名:哈特里—福克方程
- 外文名:Hartree-fock equation
- 提出者:Hartre與fock
- 套用學科:基礎物理
- 適用領域範圍:量子力學
- 適用領域範圍:多電子體系
發展簡史,導出,方程的解,套用,
發展簡史
為了解決多電子體系薛丁格方程近似求解的問題,量子化學家道格拉斯·哈特里在1928年提出了哈特里假設,將每個電子看做是在其他所有電子構成的平均勢場中運動的粒子,並且首先提出了疊代法的思路。哈特里根據他的假設,將體系電子哈密頓運算元分解為若干個單電子哈密頓運算元的簡單代數和,每個單電子哈密頓運算元中只包含一個電子的坐標,因而體系多電子波函式可以表示為單電子波函式的簡單乘積,這就是哈特里方程。但是由於哈特里沒有考慮電子波函式的反對稱要求,他的哈特里方程實際上是非常不成功的。
1930年,哈特里的學生弗拉基米爾·福克和約翰·斯萊特分別提出了考慮泡利原理的自洽場疊代方程和單行列式型多電子體系波函式,這就是今天的哈特里-福克方程。但是由於計算上的困難,HF方程誕生後整整沉寂了二十年,在1950年,量子化學家克萊門斯·羅特漢想到將分子軌道用原子軌道的線性組合來近似展開,而得到了閉殼層結構的羅特漢方程。
1953年,美國的R.帕里瑟、R.帕爾和英國的約翰·波普爾花費兩年時間使用手搖計算器分別獨立地實現了對氮氣分子的RHF自洽場計算,這是人類首次通過求解HF方程獲得對化學結構的量子力學解釋,也是量子化學計算方法第一次實際完成。在第一次成功之後,伴隨著電腦技術的迅猛發展,HF方程與量子化學一道獲得長足發展,在HF方程的基礎上,人們發展出了高級量子化學計算方法,使得計算精度進一步提高,通過對HF方程電子積分的簡化和參數化,人們大大縮減了量子化學的計算量,使得對超過1000個原子的中等大小分子的計算成為可能。
導出
哈特里-福克方程源於對多電子體系電子波函式的變分法處理。在玻恩-奧本海默近似條件下,一個多電子體系的電子運動與能量可以與原子核的運動和能量相互分離,這樣利用電子哈密頓算符和多電子波函式便可以計算體系的電子能量。其能量的表達式為:




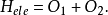





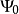



變分法的處理過程如下:令






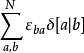







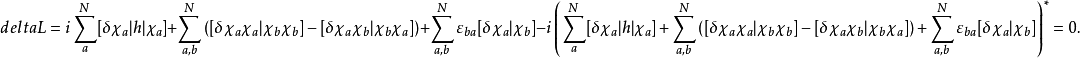

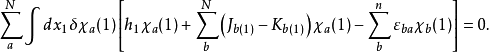






此方程形式上為本徵方程,但是福克算符中的庫侖算符和交換算符都與分子軌道有關,因此只能夠通過自洽疊代的方法近似求解,即哈特里-福克自洽場(HF-SCF)方法。HF-SCF方法是組態相互作用方法、多體微擾理論、半經驗量子化學計算等現代量子化學計算方法的基礎。
方程的解
正則哈特里-福克方程雖然具有簡單的本徵方程形式,但福克運算元中的庫侖運算元和交換運算元中含有所有 的表達式,因而方程的實際形式非常複雜,無法求得精確的解析解,只能使用疊代法求解,即量子化學中的自洽場方法。
的表達式,因而方程的實際形式非常複雜,無法求得精確的解析解,只能使用疊代法求解,即量子化學中的自洽場方法。



最終HF方程的形式轉化為廣義本徵方程

此方程稱為哈特里-福克-羅特漢方程,或羅特漢方程。這樣,相對複雜的本徵方程就轉化為只需要進行簡單代數計算就可以求解的矩陣本徵方程,而原方程中複雜的積分則在上述轉化過程中一次性完成了。
求解時首先需要給定一個分子軌道的初始猜測C,用這套分子軌道計算福克矩陣F,再求解哈特里-福克-羅特漢方程,得到一組新的分子軌道C和相應的本徵值矩陣 ,再使用新軌道C計算新的福克矩陣F……重複上述過程,直到C的各個矩陣元數值不再有明顯的變化,稱作自洽疊代收斂,就得到了HF方程的解。
,再使用新軌道C計算新的福克矩陣F……重複上述過程,直到C的各個矩陣元數值不再有明顯的變化,稱作自洽疊代收斂,就得到了HF方程的解。
得到收斂的C矩陣後,將這些係數代入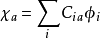 與基函式結合,便獲得了最終的分子軌道波函式以及體系電子總能量等各種性質。
與基函式結合,便獲得了最終的分子軌道波函式以及體系電子總能量等各種性質。
套用
HF方程在量子化學中有著廣泛的套用,所有分子軌道理論的量子化學計算都是以HF方程為基礎的。
- 組態相互作用方法(CI):在CI方法中,通過HF方程解得的一系列分子軌道用於構建多電子基函式集,在構建了多電子基函式集後再通過變分法處理獲得CI能量的最低點,因而進行CI計算必須首先完成HF方程的求解。
- 半經驗量子化學計算:半經驗量子化學計算是對HF方程求解過程的簡化。在HF方程的求解中,絕大部分計算量都分布在由正則HF方程向矩陣本徵方程形式轉變的過程中,如果將這一過程中大量的電子積分用經驗數值代替,便可以極大地縮短HF方法的計算時間。為此,針對不同的研究體系,量子化學家開發了不同的積分經驗常數,與之相應地產生了各種半經驗量子化學計算方法。本質上講,半經驗計算仍然是通過自洽場方法求解HF方程的過程。
