
基本介紹
- 中文名:誘導反應
- 外文名:inductive effect
- 別稱:誘導效應
- 提出者:美國化學家 路易斯
- 時間:1923年
- 對應:共價鍵的兩個原子
- 對象:電子效應
由來及意義,靜態誘導效應,- I 效應與+ I 效應,- I 效應與+ I 效應的相對強度,- I 效應與+ I 效應的酸鹼性,動態誘導效應,共價鍵的極化度,動態誘導效應的相對強度,誘導反應在有機反應中的套用,親電加成反應的相對速度,親電取代反應,誘導效應對反應產物的影響,
由來及意義
早在1861年,布特列洛夫就指出,原子在分子中能直接或間接地相互影響,這種影響是決定分子性質的重要因素。從這個觀點出發,布特列洛夫進一步發展了結構學說,說明分子性質對化學結構的依賴關係,從而奠定了有機化學的理論基礎。
1916年路易斯曾提出共價鍵的學說,他認為當原子達到稀有氣體原子結構(最外層都是含2和8個電子)的結構趨向時,可以由兩個原子共享一對電子來完成,是共價單鍵就是兩個原子間的一對共享電子所構成的一種聯繫。這個共價鍵的電子對概念迅速為化學界所一致接受。但是為什麼兩個原子間可以共享一對電子以構成一個固定的共價鍵,以及共價鍵為什麼有固定的安定性和鍵能、鍵長等性質,則未得到解釋。
從1927-1931年間,經海特勒、王守競等直到1931年鮑林(Pauling) 引入電子軌道雜化理論的概念,使范霍夫(Vant Hoff)等的碳的正四面體形象學說,可以由量子力學的理論推衍出來;另一方面,由於認識了雜化方式(SP、SP2、SP3)的不同,也就認識了形成的共價鍵又有σ鍵與π鍵之分。這樣就使我們對於單鍵、雙鍵及三鍵的特性有了正確的認識,從而對化學鍵本性的理論增添了新鮮內容。
由於對共價鍵本性的闡明,基團相互影響理論也得到進一步發展,其中首先得到解釋的就是誘導效應。1923年路易斯(Lewis)認為誘導效應是由一個電負性較強的原子X代替碳上氫原子以後,在C一X鍵上產生一個極性效應,由於鍵上的這個極性,通過電性的誘導作用,就在分子中其他鍵上引起一系列的極性變化,結果在整個分子中引起一個向著X原子方向的大規模的電子運動,這種電子運動稱為誘導效應。

正電性的原子也可以在分子中引起一系列的極性變化, 只是所產生的誘導效應的方向正好相反。
誘導效應有靜態誘導效應與動態誘導效應兩種。由極性鍵所表現出的誘導效應稱做靜態誘導效應,而在化反應過程中由於外電場(如試劑、溶劑)的影響所產生的極化鍵所表現出的誘導效應稱做動態誘導效應。誘導效應只改變鍵內電子云密度分布,而不改變鍵的本性。且與共軛效應相比,無極性交替現象。
靜態誘導效應
靜態誘導效應是由於分子本身結構決定的,與外電場(極性溶劑或試劑) 的存在無關。這種由於分子本身所固有的極化效應稱為靜態誘導效應。這種效應是永久性的,它沿著σ鍵傳遞,但隨碳鏈的增長而逐漸減弱或消失,一般傳遞到第三個碳原子就微弱了,可略而不計。
- I 效應與+ I 效應
關於分子在靜止狀態的電子云密度的分布以及此而產生的性質, 原因在於成鍵原子或基團的電負性不同, 使分子中成鍵原子的電子云發生轉移, 轉移的程度是由成鍵原子或基團的相對電負性來決定的。
靜態誘導效應通常採用烷烴H一CR3上的氫作為比較標準。

用飽和的H一CR3鍵的誘導效應為比較標準,規定為零。如果用X取代了H一CR3中氫原子後,化合物X一CR3部分的電子云密度比在H一CR3中小。X叫做吸電子基團,由吸電子基團引起的誘導效應,叫做吸電子誘導效應,常用- I 來表示。如果用Y取代H一CR3中的氫原子後,化合物Y一CR3中CR3部分的電子云密度比在H一CR3中大。Y就叫做斥電子基團,由斥電子基團引起的誘導效應,叫做斥電子誘導效應,常用+ I 來表示。
- I 效應與+ I 效應的相對強度
靜態誘導效應的強弱,決定於取代基的吸電子或斥電子能力,因此與它們的電負性有關。
在H左邊的是吸電子基團,在H 右邊的是斥電子基(供電基),離H愈遠則吸電子和斥電子的能力越大,由它所產生的誘導效應也愈強。
對同族元素來說,吸電子誘導效應:
一F > 一C l > 一B r > 一I
對同周期元素來說,吸電子誘導效應:
一F > 一OR > 一NR2
對不同雜化狀態的碳原子來說,S成分多,吸電子能力強。
一N > =NR > 一NR2
一C=CR > 一C6H5 > 一CR=CR2 > 一CR2一CR2
具有斥電子效應的基團,主要是烷基,其相對強度如下:
(CH3)3C一>(CH3)2CH 一>CH3CH2 一> CH3一
烷基只與不飽和碳相連時才呈+ I 效應。
- I 效應與+ I 效應的酸鹼性
原子或取代基的電負性,通常是由測定取代酸的電離常數而求出。如未取代的醋酸,是一種弱酸,它的離解是根據下面化學平衡來進行的:

當α位上引入一個電負性比氫強的氯原子,就誘導了電子對沿著分子中原子鏈向著氯原子轉移,結果增進了輕基(一OH)中氫原子離解成質子的傾向。所產生的氯乙酸成為較強的酸,其電離常數(Ka=155*10^-5),比醋酸約大八十多倍。

若用氯原子繼續取代α位上的氫原子,所得產物的酸性也將繼續增強。
動態誘導效應
在化學反應中, 分子反應中心如果受到極性試劑的進攻,則鍵的電子云分布將受到試劑電場的影響而發生變化。這種改變與外界電場強度及鍵的極化能力有關。分子在試劑電場影響下所發生的誘導極化,是一種暫時的現象,只有在進行化學反應的瞬間才表現出來,這種誘導效應稱為動態誘導效應。
共價鍵的極化度
分子在電性方面的表現,是因誘導效應而引起電子云的密度重新分布,使分子表現出一定的極化度。鍵的誘導極化(m)是與外界電場強度(F)成正比的。
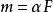
α為鍵的極化度。假定電場強度不變,哪么各種鍵的誘導極化,均由它的極化度來袂定。鍵的極化度主要是依賴於鍵內電子的流動性,也可由鍵的折光率推算出來。由於鍵的極化度是一種暫時的性質,只有在外電場影響下才表現出來。而這種外電場並不是特別布置的,溶劑的極性和反應試劑的極性都足以誘發。從內因方面考慮,動態誘導效應的大小,與成鍵元素原子核對其所屬價電子的控制能力有關,價電子離核越近,核對它的控制能為越大,則動態效應越小,因為價電子被原子核束縛住了,不易導致電子云密度的重新分布。反之,價電子離原子核越遠。核對它的控制能力就越小,則動態誘導效應也就越大。
動態誘導效應的相對強度
在外電場相同的條件下,動態誘導效應既與鍵的極化度有關,那么從元素電負性來說,電負性較大的元素對於電子的約束力也較強,因此,同一族的元素,它們的動態誘導效應隨原子序數增加而增加,對同一周期的元素, 則因原子序數的增加而降低。
I > Br > CI > F
CR3 > NR2 >OR > R > F
誘導反應在有機反應中的套用
親電加成反應的相對速度
乙烯及其衍生物與澳的加成反應是親電加成,因此試劑急是首先向電子云密度大的地方進攻。這樣在C=C 上連有+ I 基團時,將增加C=C 上π電子云的密度,從而有利於親電試劑的進攻,使反應速度加快,所以四甲基乙烯的溴化反應速度最大。反之,在C=C上若連有- l 基團時,(如一Cl、一COOH等)能降低C=C 上的π電子云,從而不利於親電試劑的進攻,因而反應速度大大降低。
親電取代反應
從反應歷程看,親電取代反應,都屬於共價鍵異裂的離子型反應。反應時親電試劑(進攻試劑)首先形成正離子,然後這個正離子向苯環上電子云密度大的位置進攻。假若由於引入取代基而增加苯環上的電子云密度,也就相應地增加親電反應的速度。在親電取代反應中,反應物通常作為電子給予體,即提供電子以形成新鍵。而進攻試劑則作為電子接受體以形成新鏈。在上述反應中,苯環就是電子的給予體,進攻試劑Cl2、HNO3、H2SO4、鹵代烷、醯鹵等都是電子接受體。
誘導效應對反應產物的影響
誘導效應對於反應的影響,在芳香族化合物的取代反應中表現得最為明顯。一般說來,鄰位對位定位基(除鹵素外)都是供電子的基團,能使苯環上的電子云密度增加,有利於親電取代,故使苯環活化。而間位定位基都是些吸電子的基團,使苯環上的電子云密度降低,不利於親電取代,故使苯環鈍化。由於苯環是一個閉合的共扼體系,若環上連有第一類定位基,定位基的影響沿著共扼鏈傳遞時,則共扼鏈上出現電子云密度大小交換,結果使鄰、對位上的電子云密度增大。若環上連有第二類定位基,影響結果則使間位上的電子云密度相對於鄰、對位來講,稍大一些。
